
bio_hansel
This workflow uses the following software for the bio_hansel pipeline. The pipeline requires the following tool:
| Tool Name | Owner | Tool Revision | Toolshed Installable Revision | Toolshed |
|---|---|---|---|---|
| bio_hansel | nml | 4654c51dae72 | 9 (2018-05-09) | Galaxy Main Shed |
To install these tools please proceed through the following steps.
Step 1: Galaxy Conda Setup
Galaxy makes use of Conda to automatically install the dependencies for bio_hansel. Please verify that the version of Galaxy is >= v16.01 and that you have conda version >= 4.3. You can upgrade conda using conda update conda. Make sure that Galaxy has been setup to use conda by modifying the appropriate configuration settings, see here for additional details.
Step 2: Install Galaxy Tools
Please install the tools as mentioned in the table above by logging into Galaxy, navigating to Admin > Search and browse tool sheds, searching for the appropriate Tool Name and installing the appropriate Toolshed Installable Revision.
The install progress can be checked by monitoring the Galaxy log files galaxy/*.log. On completion you should see a message of Installed next to the tool when going to Admin > Manage installed tool shed repositories.
Step 3: Testing the Pipeline
A Galaxy workflow and some test data has been included with this documentation to verify that all tools are installed correctly. To test this pipeline, please proceed through the following steps.
- Upload the bio_hansel Galaxy Workflow by going to Workflow > Upload or import workflow.
-
Upload the sequence reads by going to Analyze Data and then clicking on the upload files from disk icon
 . Select the test/reads files. Make sure to change the Type of each file from Auto-detect to fastqsanger. When uploaded you should see the following in your history.
. Select the test/reads files. Make sure to change the Type of each file from Auto-detect to fastqsanger. When uploaded you should see the following in your history.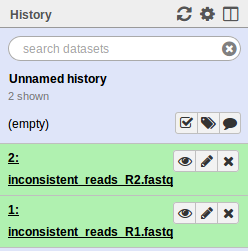
-
Construct a dataset collection of the paired-end reads by clicking the Operations on multiple datasets icon
 . Please check off the two .fastq files and then go to For all selected… > Build List of dataset pairs. You should see a screen that looks as follows.
. Please check off the two .fastq files and then go to For all selected… > Build List of dataset pairs. You should see a screen that looks as follows.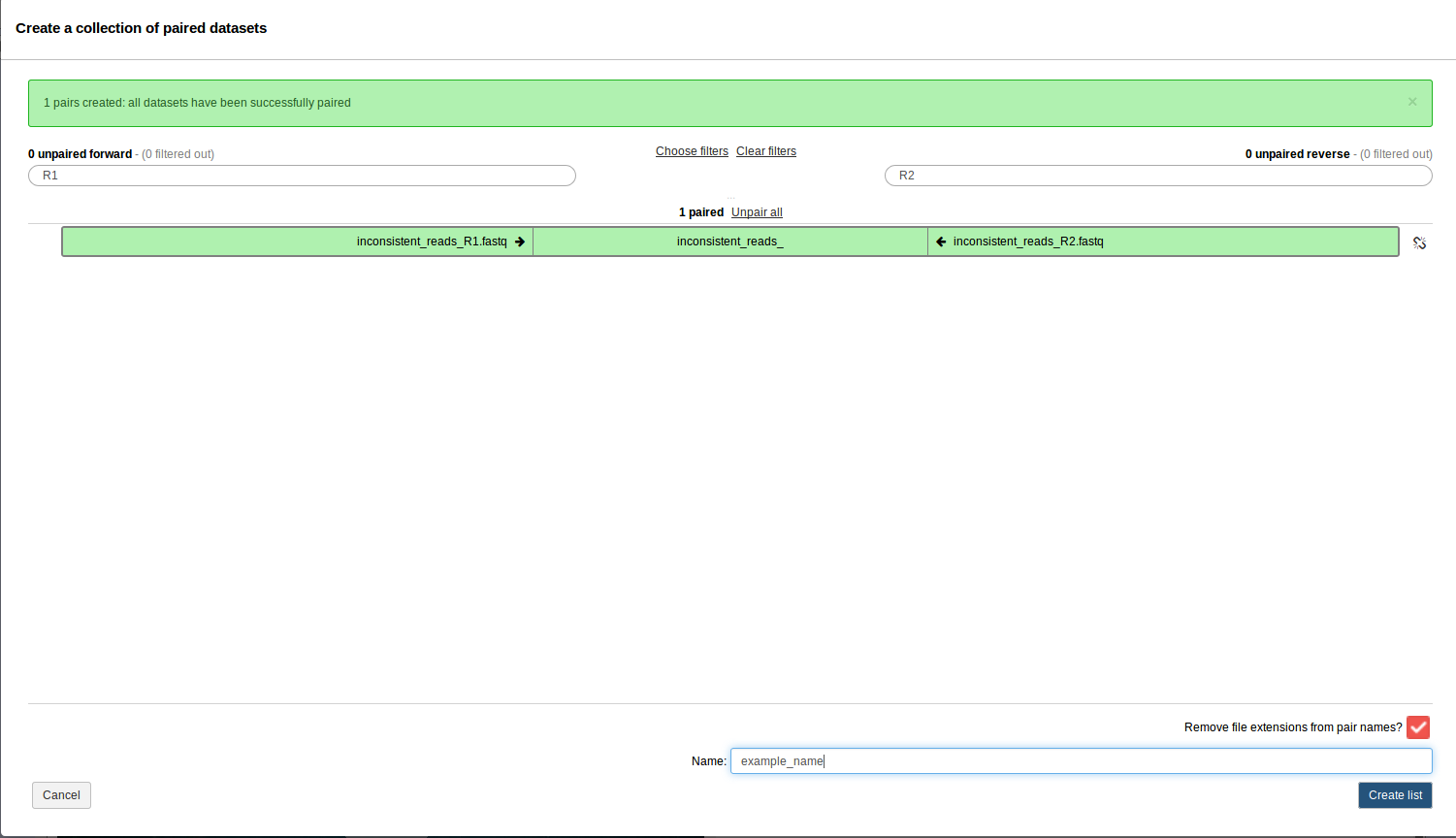
- This should have properly paired your data. Enter the name of this paired dataset collection at the bottom and click Create list.
- Run the uploaded workflow by clicking on Workflow, clicking on the name of the workflow bio_hansel (imported from uploaded file) and clicking Run. This should auto fill in the dataset collection. At the very bottom of the screen click Run workflow.
-
If everything was installed correctly, you should see each of the tools run successfully (turn green). On completion this should look like.
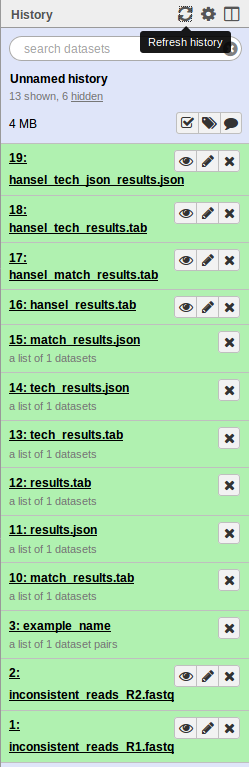
If you see any tool turn red, you can click on the view details icon
 for more information.
for more information.
If everything was successful then all dependencies for this pipeline have been properly installed.