
IRIDA Workflow Description
This file describes the IRIDA Workflow Description format. This is an XML format that is used to link up a Galaxy Workflow and the corresponding Galaxy Tools into IRIDA and provide IRIDA with the necessary information on the input datasets for Galaxy, parameters, and output files to save.
- IRIDA Workflow Description
- Workflow Description XML Element Details
<inputs>Elements<parameters>Elements<outputs>Elements<toolRepositories>Elements- Workflow Description Example
Workflow Description XML Element Details
<iridaWorkflow>
The outer-most element tag. All other tags are contained within an <iridaWorkflow>.
Attributes
None.
Example
<iridaWorkflow>
...
</iridaWorkflow>
<id>
An ID for the workflow which is used to uniquely identify this exact workflow. This should be a UUID. A quick way to generate a random (version 4) UUID is with the uuid command, as in uuid -v 4.
Attributes
None.
Example
<id>79872edb-4f3b-4a51-aff7-33ddbc05ec5e</id>
<name>
The name of the workflow.
Attibutes
None.
Example
<name>MyPipeline</name>
<version>
A version number for the workflow. This should be incremented on every modification to the workflow.
Attributes
None.
Example
<version>1.0</version>
<analysisType>
The particular type or class of analysis this workflow belongs to. This must be one of the types defined in the AnalysisType enum in IRIDA. Currently, the type can be either assembly-annotation or phylogenomics. Please see the AnalysisType JavaDoc for more details.
Attributes
None.
Example
<analysisType>phylogenomics</analysisType>
<inputs>
A description of the different types of inputs to this workflow. This must contain at least one of <sequenceReadsPaired> or <sequenceReadsSingle> as a sub-element.
Attributes
None.
Example
<inputs>
<sequenceReadsPaired>sequence_reads_paired</sequenceReadsPaired>
</inputs>
<parameters>
This defines the set of parameters for a workflow as well as how to map these parameters to Galaxy tool names and versions.
Attributes
None.
Example
<parameters>
<parameter name="myparameter" defaultValue="HKY85">
<toolParameter toolId="irida.corefacility.ca/galaxy-shed/repos/irida/phyml/phyml1/3.1"
parameterName="datatype_condition.model" />
</parameter>
</parameters>
<outputs>
Used to define a list of output files from the Galaxy workflow which will be saved back into IRIDA.
Attributes
None.
Example
<outputs>
<output name="tree" fileName="phylogeneticTree.tre" />
</outputs>
<toolRepositories>
Defines a list of the repositories storing the dependency Galaxy tools for this workflow. This is used to keep track of the particular versions of dependencies and can also be used for integration testing with Galaxy.
Attributes
None.
Example
<toolRepositories>
<repository>
<name>spades</name>
<owner>lionelguy</owner>
<url>https://toolshed.g2.bx.psu.edu/</url>
<revision>21734680d921</revision>
</repository>
</toolRepositories>
<inputs> Elements
<sequenceReadsPaired>
An optional element tag contained in the <inputs> tag set. This defines the name, if any, of a list of paired-end files used as input to the Galaxy workflow.
Attributes
None.
Example
If a Galaxy workflow contains an Input dataset collection of type list:paired then the name of the dataset collection, sequence_reads_paired, should correspond to the name in this element. Please see the image below.
Galaxy Workflow Input
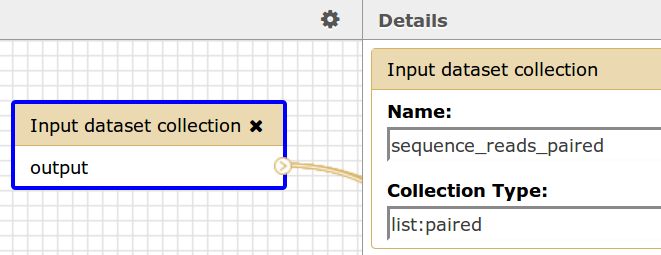
Workflow Description File
<sequenceReadsPaired>sequence_reads_paired</sequenceReadsPaired>
<sequenceReadsSingle>
An optional element tag contained in the <inputs> tag set. This defines the name, if any, of a list of single-end files used as input to the Galaxy workflow.
Attributes
None.
Example
If a Galaxy workflow contains an Input dataset collection of type list then the name of the dataset collection, sequence_reads_single, should correspond to the name in this element. Please see the image below.
Galaxy Workflow Input
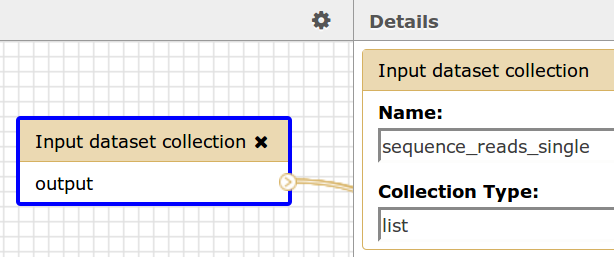
Workflow Description File
<sequenceReadsSingle>sequence_reads_single</sequenceReadsSingle>
<reference>
An optional element tag contained in the <inputs> tag set. This defines the name, if any, of the reference input for the Galaxy workflow.
Attributes
None.
Example
If a Galaxy workflow contains an Input dataset for a reference genome, then the name of the dataset, reference, should correspond to the name in this element. Please see the image below.
Galaxy Workflow Input
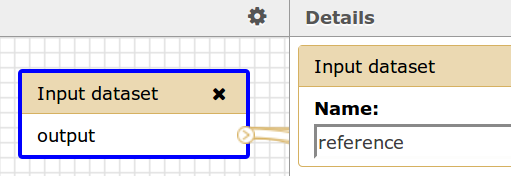
Workflow Description File
<reference>reference</reference>
<requiresSingleSample>
An optional element tag contained in the <inputs> tag set. This defines whether or not this workflow only operates on a single sample true or can handle multiple samples false. That is to say, if this workflow will upload only a single sample to Galaxy to execute the workflow, such as with the assembly and annotation workflow, then this should be set to true. Otherwise this should be set to false. The default is false.
Attributes
None.
Example
<requiresSingleSample>false</requiresSingleSample>
<parameters> Elements
<parameter>
Contained in the <parameters> element tag. This defines a single parameter for a workflow. This must contain at least one <toolParameter> element which defines the specific Galaxy tool and parameter to override. The defaultValue should also correspond to one of the acceptible Galaxy parameter values. If no defaultValue can be specified, then the required attribute should be set to true, indicating that the user must input a parameter value before each pipeline run.
Attributes
| attribute | type | details | required | example |
|---|---|---|---|---|
| name | string | The name of the parameter. This will be used in the IRIDA database and configuration files to refer to this parameter. | yes | name="myparameter" |
| defaultValue | string | The default value of the parameter. | yes | defaultValue="1" |
| required | boolean | “true” if no default value can be set for the parameter, so the user must select a value before launching. Defaults to “false” if not set. | no | required="true" |
Example
To override the model parameter defined in the Galaxy version of PhyML in https://github.com/phac-nml/snvphyl-galaxy/blob/v0.1/tools/phyml/phyml.xml, the following entry can be used. The defaultValue="HKY85" must correspond to the value defined in the Galaxy PhyML Tool (see phyml.xml#L38).
<parameter name="myparameter" defaultValue="HKY85">
<toolParameter toolId="irida.corefacility.ca/galaxy-shed/repos/irida/phyml/phyml1/3.1"
parameterName="datatype_condition.model" />
</parameter>
<dynamicSource>
Attributes
(None)
<galaxyToolDataTable>
Attributes
| attribute | type | details | required | example |
|---|---|---|---|---|
| name | string | Name of the Galaxy Tool Data Table from which parameter values will be pulled. | yes | name="mentalist_databases" |
| displayColumn | string | The name of the Tool Data Table column containing a human-readable label for the parameter value. | yes | displayColumn="name" |
| parameterColumn | string | The name of the Tool Data Table column containing the parameter value to be passed to the pipeline. | yes | parameterColumn="value" |
Example
Dynamic sources are used to pull parameter values from outside systems such as Galaxy Tool Data Tables
<parameter name="kmer_db" required="true">
<dynamicSource>
<galaxyToolDataTable name="mentalist_databases" displayColumn="name" parameterColumn="value" />
</dynamicSource>
<toolParameter toolId="toolshed.g2.bx.psu.edu/repos/dfornika/mentalist/mentalist_call/0.1.3"
parameterName="kmer_db" />
</parameter>
<toolParameter>
Contained in the <parameter> element tag. This defines a parameter in a Galaxy tool to map to from the parent <parameter> definition. The toolId and parameterName must correspond to the information defined in the Galaxy workflow and Galaxy tool XML configuration files.
Attributes
| attribute | type | details | required | example |
|---|---|---|---|---|
| toolId | string | The id of the tool in the Galaxy workflow of the parameter to override. | yes | toolId="my_galaxy_tool" |
| parameterName | string | The name of the parameter in the Galaxy tool to override. | yes | parameterName="parameter.section.name" |
Example
For the tool PhyML in the SNVPhyl Galaxy Workflow, the tool_id is given below:
tool_id (see snvphyl-workflow.ga#L506)
"tool_id": "irida.corefacility.ca/galaxy-shed/repos/irida/phyml/phyml1/3.1"
This corresponds to the toolId attribute to use in the <toolParameters> element (tool_id="irida.corefacility.ca/galaxy-shed/repos/irida/phyml/phyml1/3.1").
For the model parameter name, the corresponding line in the SNVPhyl Galaxy workflow is given below:
model (see snvphyl-workflow.ga#L507)
"tool_state": "...\\\"model\\\": \\\"HKY85\\\"...
However, this parameter is stored as a sub-element of other parameters. To get the full parameter name the PhyML Galaxy Tool XML file must be referenced:
parameterName (see phyml.xml#L6)
<command interpreter="bash">./phyml.sh ... -m $datatype_condition.model ...
...
</command>
Since the string datatype_condition.model is used to refer to the model parameter, this should be used as the full parameterName attribute.
Putting both these together into the IRIDA Workflow Description <toolParameter> element gives:
<toolParameter toolId="irida.corefacility.ca/galaxy-shed/repos/irida/phyml/phyml1/3.1"
parameterName="datatype_condition.model" />
<outputs> Elements
<output>
Contained in the <outputs> element tag. Defines a particular output file from a Galaxy workflow. In order to save this output file in IRIDA, the name of the dataset file in Galaxy must be defined in the fileName attribute of the <output> element. This can be accomplished by using the Rename Dataset action in a Galaxy workflow to give an output dataset file an explicit name.
Attributes
| attribute | type | details | required | example |
|---|---|---|---|---|
| name | string | A label for the output file name. This is used internally to map to a particular output file in the IRIDA database. | yes | name="my-output-1" |
| fileName | string | The name of an output file from the Galaxy workflow. This is used to find the particular output file when transferring results back to IRIDA. | yes | fileName="output-file.txt" |
Example
If the Rename Dataset action is set on a tool to give an output file the name phylogeneticTree.tre
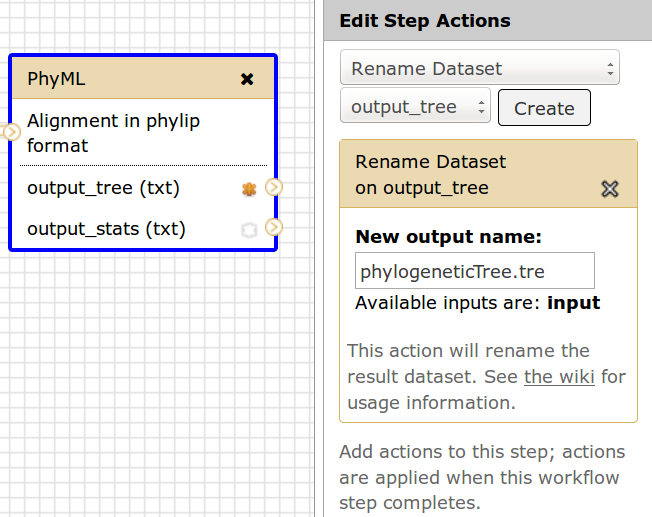
then the corresponding dataset will appear with this name in the Galaxy History.

This name must then correspond to the fileName in the workflow description file.
Workflow Description File
<output name="tree" fileName="phylogeneticTree.tre" />
<toolRepositories> Elements
<repository>
Contained within a <toolRepositories> element tag. Defines a particular repository storing a dependency for the workflow. This must correspond to the information within some Galaxy ToolShed containing this repository. For example, for SPAdes https://toolshed.g2.bx.psu.edu/view/lionelguy/spades/21734680d921.
Attributes
None.
Example
For the version of SPAdes stored within the main Galaxy ToolShed, https://toolshed.g2.bx.psu.edu/view/lionelguy/spades/21734680d921, the following information should be defined.
<repository>
<name>spades</name>
<owner>lionelguy</owner>
<url>https://toolshed.g2.bx.psu.edu/</url>
<revision>21734680d921</revision>
</repository>
<name>
Contained in the <repository> element tag. Defines the name of the tool repository.
Attributes
None.
Example
For SPAdes, https://toolshed.g2.bx.psu.edu/view/lionelguy/spades/21734680d921, the name would be spades.
<name>spades</name>
<owner>
Contained in the <repository> element tag. Defines the owner of the tool repository.
Attributes
None.
Example
For SPAdes, https://toolshed.g2.bx.psu.edu/view/lionelguy/spades/21734680d921, the owner would be lionelguy.
<owner>lionelguy</owner>
<url>
Contained in the <repository> element tag. Defines the url of the tool repository.
Attributes
None.
Example
For SPAdes on the main Galaxy ToolShed, https://toolshed.g2.bx.psu.edu/view/lionelguy/spades/21734680d921, the url would be https://toolshed.g2.bx.psu.edu/.
<url>https://toolshed.g2.bx.psu.edu/</url>
<revision>
Contained in the <repository> element tag. Defines the revision number of tool.
Attributes
None.
Example
For the version of SPAdes on the main Galaxy ToolShed, https://toolshed.g2.bx.psu.edu/view/lionelguy/spades/21734680d921, the revision would be 21734680d921.
<revision>21734680d921</revision>
Workflow Description Example
An example workflow description XML file is given below.
<?xml version="1.0" encoding="UTF-8"?>
<iridaWorkflow>
<id>79872edb-4f3b-4a51-aff7-33ddbc05ec5e</id>
<name>MyPipeline</name>
<version>1.0</version>
<analysisType>phylogenomics</analysisType>
<inputs>
<sequenceReadsPaired>sequence_reads_paired</sequenceReadsPaired>
<reference>reference</reference>
<requiresSingleSample>false</requiresSingleSample>
</inputs>
<parameters>
<parameter name="myparameter" defaultValue="HKY85">
<toolParameter toolId="irida.corefacility.ca/galaxy-shed/repos/irida/phyml/phyml1/3.1"
parameterName="datatype_condition.model" />
</parameter>
</parameters>
<outputs>
<output name="tree" fileName="phylogeneticTree.tre" />
</outputs>
<toolRepositories>
<repository>
<name>spades</name>
<owner>lionelguy</owner>
<url>https://toolshed.g2.bx.psu.edu/</url>
<revision>21734680d921</revision>
</repository>
<repository>
<name>phyml</name>
<owner>irida</owner>
<url>https://irida.corefacility.ca/galaxy-shed</url>
<revision>b5867c5c7674</revision>
</repository>
</toolRepositories>
</iridaWorkflow>