
Using the SISTR Pipeline
This guide describes how to make use of the Salmonella in-silico Typing Resource (SISTR) pipeline within IRIDA. This pipeline enables the identification of the serovar and cgMLST types for Salmonella whole genome sequencing (WGS) data through comparisons with a large database (10,000+) of Salmonella genomes within NCBI.
Pipeline Overview
The SISTR pipeline that is implemented within IRIDA makes use of the following steps to translate WGS data into typing information:
- Paired-end reads are merged with FLASH.
- Merged and un-merged reads are assembled de novo using SPAdes.
- Low-coverage and small contigs are removed from the generated assembly.
- The assembled genome is passed to sistr_cmd, a command-line program for comparing genomes against the SISTR database.
Running the Pipeline
The SISTR pipeline can bet set up to run using two separate methods.
1. Automated Execution
The SISTR pipeline can be set to run automatically on upload of new sequencing data to particular projects.
Project Settings
Automated SISTR analysis can also be enabled (or disabled) after a project is created from the project Settings page.
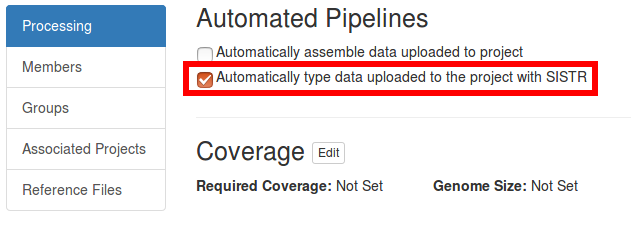
If automated execution of SISTR has been enabled for a project, then a SISTR analysis will be scheduled for execution on the upload of new sequencing data. The results are accessible from the particular Project > Analyses page.
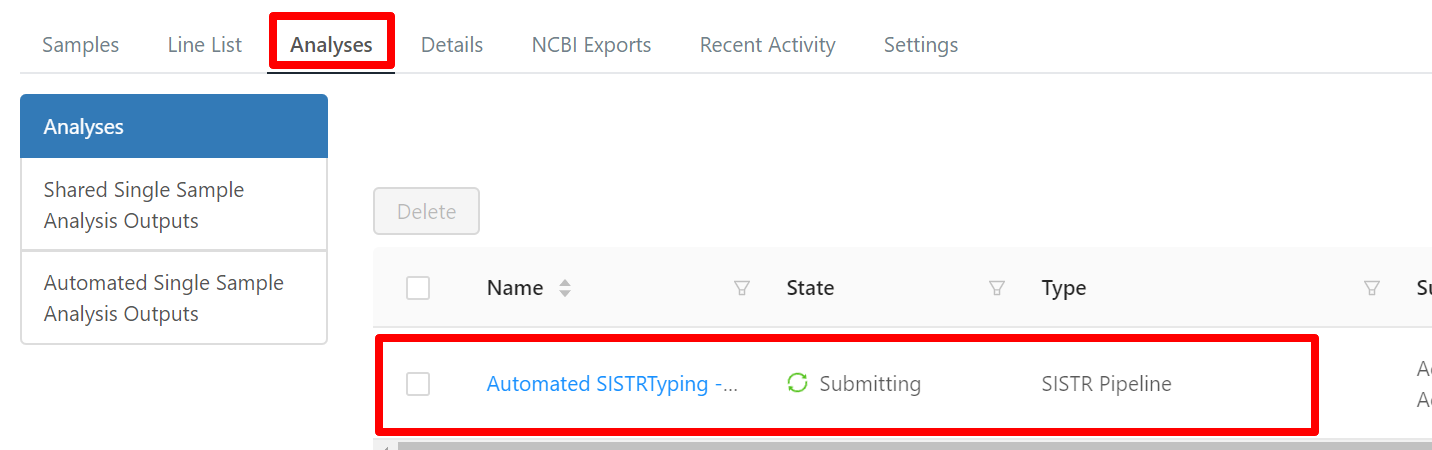
Clicking the Automated SISTR Typing link brings you to the appropriate analysis page for SISTR.
2. Manual Execution
To execute SISTR manually, please refer to the IRIDA/SISTR Tutorial.
SISTR Results
A successful SISTR run should produce the following page as output. There are three possible Quality Control Statuses (Pass, Warning, and Fail)
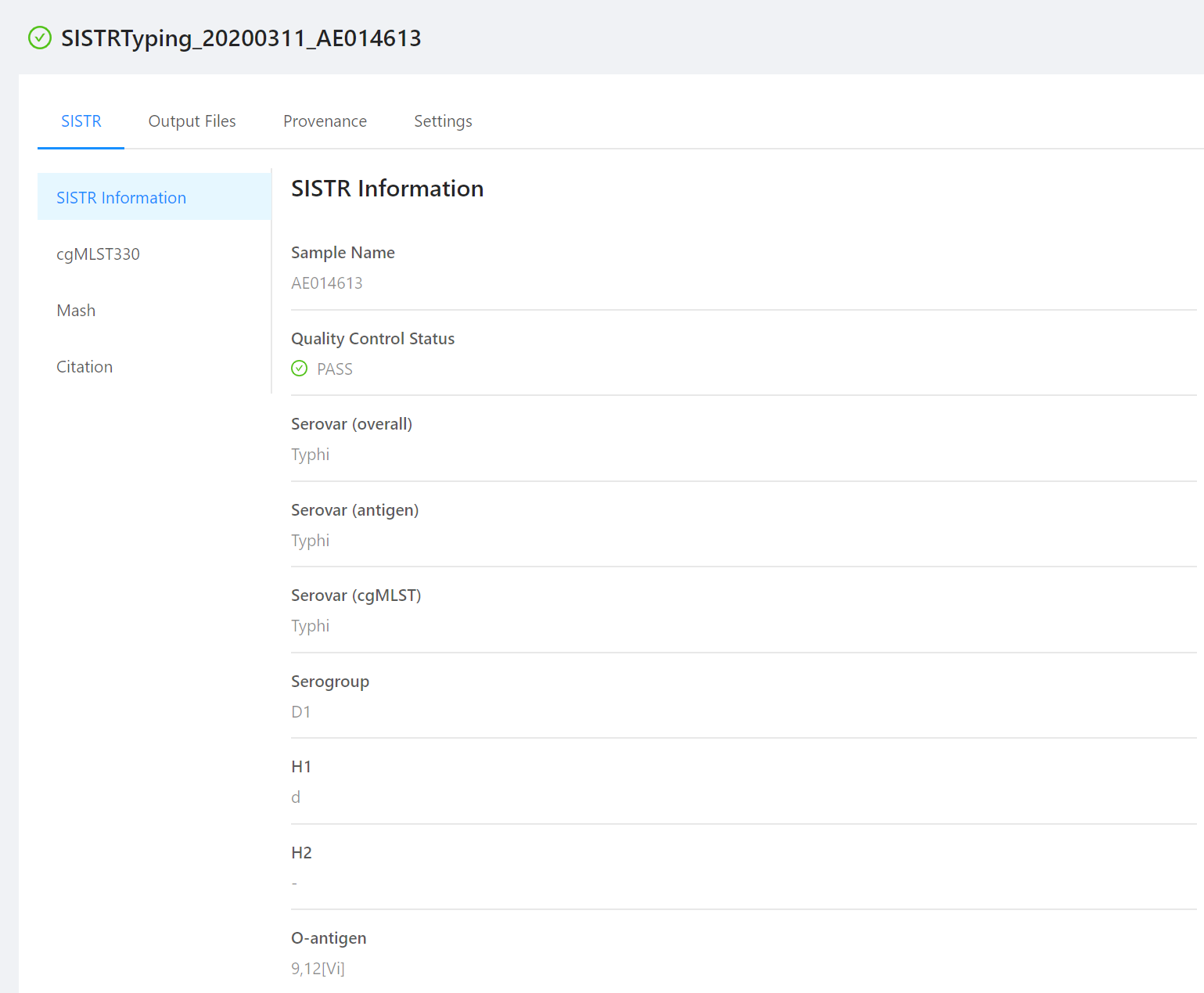
The results are broken up into three different sections (SISTR Information, cgMLST330, and Mash).
To view the output files and/or download the outputs, click the Output Files tab.
Report
Interpretation of the produced output is as follows:
1. SISTR Information
Basic information on the sample and quality of the SISTR results.
- Sample Name: The name of the sample used within this analysis.
- Quality Control Status: A value of
PASS,FAIL, orWARNINGdepending on the quality of the input genome as determined by SISTR. - Quality Control Details: In the case of a status of
WARNINGorFAIL, the particular reason why these results did not pass.
Serovar Predictions:
The in silico serovar predictions generated from SISTR.
- Serovar (overall): The overall serovar prediction from both the antigen and cgMLST methods employed by SISTR.
- Serovar (antigen): The serovar prediction generated by looking for the presence of specific antigen genes.
- Serovar (cgMLST): The serovar prediction generated by comparing the cgMLST type of this genome to the SISTR database.
- Serogroup: The serogroup, identified based on the detected alleles of the wzx and wzy genes.
- H1: The H1 antigen prediction based off of the detected fliC allele. If absent, a
-is reported. - H2: The H2 antigen based of off the detected fljB allele. If absent, a
-is reported. - O-antigen: The O-antigen prediction (inferred from the overall serovar prediction).
2. cgMLST330
The results of additional predictions made using the SISTR cgMLST330 schema.
- Subspecies: The subspecies identified by comparing the genome to the cgMLST database in SISTR.
- Matching genome name: The name of the closest matching genome in the SISTR database.
- Alleles matching genome: The number of alleles that have a sufficient match (non-missing, non-partial/truncated) to the closest genome in the SISTR database.
- Percent matching: The percent of alleles perfectly matching the closest genome (e.g., 100% * Alleles matching genome/330).
- cgMLST Sequence Type: A SISTR Sequence Type number associated with the particular cgMLST profile. A sequence type will only be assigned when the full complement of 330 cgMLST genes are found (whether a perfect match or an imperfect but non-truncated match).
3. Mash
The results of predictions made through comparisons using the software Mash. Generally, cgMLST results are preferred over Mash.
- Subspecies: The subspecies identified by comparing the genome to the SISTR database.
- Serovar: The serovar identified using Mash.
- Matching genome name: The name of the closest matching genome in the SISTR database identified using Mash.
- Mash distance: The Mash distance to the closest matching genome. This approximates the mutation rate.
Output Files
In addition to the report, the SISTR pipeline produces the following files available for download.
sistr-cgmlst-profiles.csv: A comma-separated values file listing the cgMLST allelic profile for the genome.sistr-alleles-out.json: A JSON file containing details on each of the allele calls.shovill-contigs.fasta: The set of contigs generated from the de novo assembly and used by SISTR. This file could be uploaded to the SISTR web application for additional details and visualization of the results.sistr-predictions.json: The SISTR prediction results used to generate the SISTR report.sistr-novel-alleles.fasta: A list of any novel alleles that were detected by SISTR.
More information on the interpretation of these files is available on the sistr_cmd page.